![]()
Index
- Overview
- What do you need for annotating a sequence dataset?
- Tutorial
- What does Sma3s annotate?
- What's new
- References
|
New Sma3s release 2! Sma3s is now faster and more accurate, and gives suitable results for creating charts. This new release has recently published in Proteomics. Now in the directory OMICtools |
Overview
Sma3s (Sequence massive annotator using 3 modules) is an easy-to-use tool for high throughput annotation that provides both accuracy and broad applicability for different kinds of sequence datasets such as proteomes or transcriptomes.
Biological sequence annotation is the process of associating biological information to sequences of interest. Annotations can include the potential function, cellular localization, biological process or protein structure of a given sequence, and is of special interest for genomics projects, gene-expression experiments and many other emerging areas of research.
Sma3s is a tool for this important area, which is especially focused on the massive annotation of sequences from any kind of gene library or genome. It provides high levels of prediction accuracy (higher than 80%) with minimal manual input and low computational resource requirements. It is composed of three integrative modules that annotate unknown sequences with increasing difficulty. All three modules use a preliminary exhaustive BLAST search as their starting point, and together generate annotations that are both highly sensitive and specific.
Finally, Sma3s offers results in files that can be easily managed and opened with a spreadsheet program.
What do you need for annotating a sequence dataset?
Sma3s has low computing requirements and can be used on virtually any computer. It is written in Perl language and you need its interpreter (http://www.perl.com), which is preinstalled in Linux and Mac OS X (in Windows it will not be necessary). Additionally, you need to install the Blast+ package for your operating system.
Finally, you will need our Sma3s program:
>>> Sma3s_v2.pl <<< (see Creative Commons license)
To annotate your sequence dataset, you only need the following files:
- Your query sequences in multi-FASTA format,
- The reference database, which you can download from our server: http://www.bioinfocabd.upo.es/sma3s/db/
Usual command line for annotating proteomes:
./sma3s.pl -i query_dataset.fasta -d uniref90.fasta -goslim
Usual command line for annotating transcriptomes:
./sma3s.pl -i query_dataset.fasta -d uniref90.fasta -nucl -goslim
Run "sma3s_v2.pl --help" for help with these and other advanced parameters.
Alternatively, you can use Sma3s with the whole UniProt database, if you are interested in a more sensitive, though more slowly, annotation. To do that, you must download a .dat file from UniProt (ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/), and install the Blast Legacy package (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/legacy/).
Tutorial
 |
Complete tutorial to install and use Sma3s. | |
 |
Install Blast+ for Windows from: ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/ Save all the necessary files into a folder called 'annotation', and use this alternative script (instead of the above program): Sma3s_v2.exe. Execute cmd.exe, and write 'cd \annotation' in the console. Follow the above video-tutorial. |
|
 |
Install Blast+ for Mac from: ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/ Open Terminal from Applications/Utilities. Follow the above video-tutorial. |
|
What does Sma3s annotate?
UniProt is arguably the most complete publically-available protein database, and Sma3s uses several term types from this database to annotate sequences in the query dataset:
- Gene Ontology (GO). Gene Ontology provides a controlled vocabulary to describe genes and gene product attributes. It is organized into three biological ontologies: molecular function, biological process and cellular component. Sma3s offers also Slim GO terms, which represents more generic annotations.
- Swiss-Prot Keywords. The Swiss-Prot keywords constitute a well-defined and controlled vocabulary of terms used to annotate a UniProt protein entry. These keywords describe functions, biological processes, structure, cellular localization and other protein characteristics.
- Pathway annotation. This annotation provides a description of the metabolic pathway(s) in which a protein is involved. Sma3s gathers annotations at the most generic level of the hierarchy. This type of annotation is particularly useful for identifying co-expressed genes that are active in the same metabolic pathway.
New annotation types can be incorporated into Sma3s with only minor changes to its algorithms.
Sma3s sequence annotations also include both the most probable gene name and the most probable description (including putative EC enzyme codes), which can be useful for non-specialist users who only want to rapidly identify their sequences.
We have tested Sma3s with sequences from different taxonomic divisions with the following accuracy:
| Taxonomic division | Sensitivity | Specificity |
| Bacteria | 96% | 97% |
| Plants | 85% | 90% |
| Mammals | 85% | 86% |
| Invertebrates | 65% | 80% |
Output example - As an output example, you can download the following files containing an annotated bacterial proteome, which can be opened with a spreadsheet program:
File with the complete annotation:
proteome_uniref90_source_go_goslim.tsv
File with the summary, including functional categories:
proteome_uniref90_source_go_goslim_summary.tsv
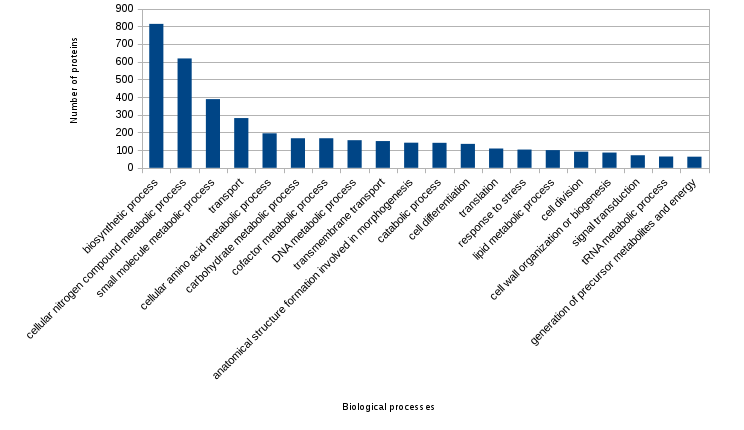
Figure created from the previous file (the most frecuent annotated biological processes):
What's new
Sma3s allows high-throughput annotation analysis to non-specialist users. These are the improvements for the new Sma3s release 2:
Performance: the algorithm is now more exhaustive and accurate
-
It offers a combined annotation from the 3 available modules, giving preference to manually curated sequences from the database.
-
Both the assigned gene name and description come from the most informative sequence.
-
The module 2 uses the Rost equation and takes in account the ortholog with the best annotation.
-
It takes automatic parameters for improving transcriptome annotations.
Usability: it requires both low knowledge and computing requirements
-
It only requires the external Blast+ package.
-
It can be easily run in Linux, Windows and Mac.
-
A reference database is provided to make easier the annotation.
Output utility: the result includes a summary useful to create figures
-
A summary of the whole annotation is given, with the number of annotated sequences and a brief about found functional classes which can be used to create article figures.
-
EC numbers are added to the annotation, and the GO terms are separately shown for each category.
-
Generic GO Slim can be added to the annotation.
-
The sequences used for the annotation can be reported, and the unannotated sequences can be discarded from the final report.
Quality: the user can select only quality predictions
-
A quality annotation can be performed by using GO and UniProt evidence codes.
-
Non-informative annotation are avoided, as well as database sequences without any GO term and UniProt keyword.
Run time: the processing time has decreased and it can be easily run in a personal computer
-
The similarity search by Blast, which is the most demanding processing time step, can be run in a multi-thread way.
-
It uses the non-redundant database UniRef90 which reduces the processing time, but keeping a high sensitivity.
-
When using UniRef90, the clustering step for removing redundancy can be avoided.
-
All of this gets a 4-fold processing increase.
References
Please cite the use of Sma3s with the last reference:
- Antonio Muñoz-Mérida, Enrique Viguera, M. Gonzalo Claros, Oswaldo Trelles, Antonio J. Pérez-Pulido (2014) Sma3s: a three-step modular annotator for large sequence datasets. DNA Research, 21(4):341-53.
- Carlos S. Casimiro-Soriguer, Antonio Muñoz-Mérida, Antonio J. Pérez-Pulido (2017) Sma3s: a universal tool for easy functional annotation of proteomes and transcriptomes. Proteomics. 17(12).
Sma3s have been widely used and cited in the literature.

Sma3s is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.